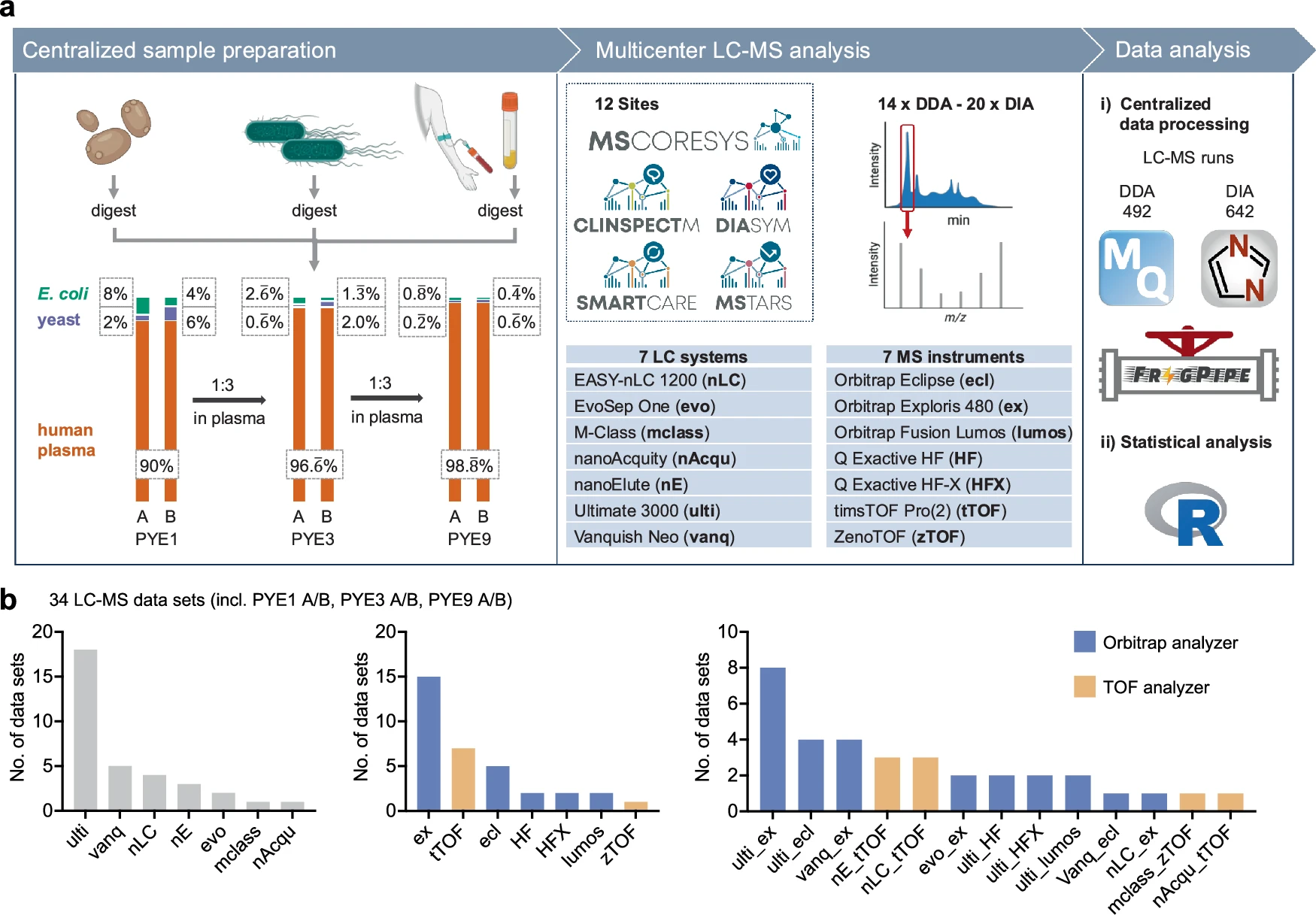
Overview of the PYE sample set and the study design. From Distler et al., 2025. “Multicenter evaluation of label-free quantification in human plasma on a high dynamic range benchmark set.“ Nat Commun 16, 8774 (2025). https://doi.org/10.1038/s41467-025-64501-z; Licensed under the terms of the Creative Commons CC-BY 4.0 license.
Human plasma holds significant clinical potential as a liquid biopsy for disease detection and monitoring. However, plasma proteome analysis using LC-MS faces a fundamental challenge: the protein dynamic range spans over 11 orders of magnitude, with highly abundant proteins like albumin (55% of total plasma protein) overwhelming detection of lower-abundance biomarkers.
Distler et al. evaluated label-free quantification performance and benchmarked standardised workflows for neat plasma analysis across twelve partner sites of the MSCoreSys clinical proteomics consortium. They developed the PYE sample set (a human tryptic plasma digest spiked with varying amounts of yeast and E. coli proteomes) to simulate physiological dynamic range while providing known protein ratios for accuracy assessment.
The study analysed the PYE samples across multiple platforms, with data acquisition employing both DDA and DIA modes. Centralised analysis used MaxQuant (DDA) and DIA-NN (DIA). Each lab used different LC-MS setups, with the Aurora Ultimate 25×75 CSI, Aurora Rapid 5×150 CSI, and Odyssey 25×75 columns being paired with timsTOF Pro and Exploris 480 mass spectrometers and with EASY-nLC 1200, nanoAcquity, and UltiMate 3000 LCs across sites A, G, and L. Ion sources included standard Nanospray Flex and CaptiveSpray configurations.
This quantitative benchmarking study revealed that DIA workflows substantially outperformed DDA approaches across all centres. DIA methods identified 3,193 proteins on average compared to 1,743 proteins with DDA – while achieving significantly higher technical reproducibility (median CV 5.9% versus 15.4% for DDA).
The study showed that modern DIA platforms reliably quantify proteins across 3–4 orders of magnitude in plasma, detecting hundreds of non‑human proteins even in the most diluted samples. This capability substantially expands the accessible biomarker space compared to historical DDA methods. The systematic analysis also revealed that chromatographic optimisation and MS hardware configuration, rather than simple gradient duration, predominantly influence proteome depth, though longer gradients generally yielded higher identification rates.
These findings establish DIA as the preferred acquisition method for clinical plasma proteomics and provide a validated benchmark resource for method standardisation and software development. The work sets a critical foundation for transitioning LC-MS proteomics from research applications to accredited clinical diagnostics, where reproducibility and standardised workflows are essential. Future clinical proteomics studies can leverage these insights to select appropriate platforms and anticipate realistic coverage depth, while the PYE dataset itself serves as a community resource for benchmarking novel analysis tools and hardware improvements as the field continues to evolve.
Publication
Nature Communications
Authors
Ute Distler, Han Byul Yoo, Oliver Kardell, Dana Hein, Malte Sielaff, Marian Scherer, Anna M. Jozefowicz, Christian Leps, David Gomez-Zepeda, Christine von Toerne, Juliane Merl-Pham, Teresa K. Barth, Johanna Tüshaus, Pieter Giesbertz, Torsten Müller, Georg Kliewer, Karim Aljakouch, Barbara Helm, Henry Unger, Dario L. Frey, Dominic Helm, Luisa Schwarzmüller, Oliver Popp, Di Qin, Susanne I. Wudy, Ludwig Roman Sinn, Julia Mergner, Christina Ludwig, Axel Imhof, Bernhard Kuster, Stefan F. Lichtenthaler, Jeroen Krijgsveld, Ursula Klingmüller, Philipp Mertins, Fabian Coscia, Markus Ralser, Michael Mülleder, Stefanie M. Hauck & Stefan Tenzer;
Title

